International Journal of Pharmaceutical and Phytopharmacological Research
ISSN (Print): 2250-1029
ISSN (Online): 2249-6084

Infectious diseases caused by viruses, bacteria, and fungi have been a significant concern for human civilization since ancient times. The intracellular proliferative features of viruses set them apart from all other infectious agents. Over the past decade, more cases of RNA virus diseases have been reported than DNA viruses. Among all RNA viruses, myocarditis-causative Coxsackie virus B3 (CVB3) grabbed the attention of drug discovery scientists due to its unique infective ability of cardiomyocytes. Due to the steep rise in myocarditis incidents, the present study was designed to screen blood-brain barrier permeable efficient Salvadora persica metabolites that can block the entry of CVB3. In the current in-silico study, we adopted Swiss ADME, trRosetta, PROCHECK, Galaxy, PyRx, and Biovia Discovery Studio to screen potent binding metabolites towards VP1 and VP2 of CVB3. During Swiss ADME analysis, we screened forty Salvadora persica metabolites and found BBB permeable methoxybenzoic acid and umbellone. While conducting molecular interaction analysis, it was also noticed that methoxybenzoic acid showed a superior binding affinity of -4.8 towards VP1, and umbellulone exhibited a binding affinity of -5.8Kcal with VP2. Based on our studies, we inferred that BBB permeable methoxybenzoic acid and umbellone are the potential phytotherapeutics to be explored in-silico, in-vitro, and in-vivo data generation, essential for the management of myocarditis.
INTRODUCTION
Infectious diseases that are caused by viruses, bacteria, and fungi have been well-known to human civilization since ancient times. Researchers were drawn to viruses because of their unique obligatory intracellular proliferative characteristics. The infections caused by corona, human immunodeficiency, nipah, hanta, herpes simplex,
chikungunya, human entero, dengue, Japanese encephalitis, influenza, chandipura, buffalopox, crimeancongo, and other viruses affected human health for the past few decades [1]. Currently, along with these existing viruses, coxsackie virus B3 (CVB3), a serotype of coxsackie virus responsible for myocarditis, has been turning into a global burden.
Myocarditis is an inflammation of the myocardium that can be caused by either infectious or non-infectious agents. It is estimated that around 1.8 million cases of myocarditis occur annually worldwide, with a global prevalence range of 10.2 to 105.6 cases per 100,000 individuals [2]. Acute myocarditis is caused by the Coxsackie B3 virus. CVB3 possesses a symmetrical icosahedral capsid that holds a positive sense RNA genome. The capsid is composed of twelve pentamers, each made up of asymmetrical units of structural proteins VP1-VP4. In order to stabilize the capsid pentamers throughout the virus assembly process, VP4, which is located on the inner surface of the shell, links the N-termini of different capsid proteins to the viral RNA. VP1, VP2, and VP3 work together to create the viral shell [3]. Among all the characteristics, the ability to bind to the Coxsackie virus and the adenovirus receptor (CAR), which are present on the transmembrane of cardiomyocytes and are crucial in coupling neighboring cardiomyocytes, attracted the interest of drug discovery scientists who wanted to create therapeutics that targeted the viral capsid subunit and infiltrated the CVB3 without harming the host biological system.
Over the past few decades, with the advent of science and technology, drug discovery scientists discovered a large number of expensive synthetic antiviral therapeutics, viz., abacavir, amprenavir, atazanavir, baricitinib, chloroquine, darunavir, didanosine, delavirdine, efavirenz, emtricitabine, favipiravir, indinavir, lamivudine, lopinavir, maraviroc, nevirapine, nelfinavir, remdesivir, ritonavir, stavudine, saquinavir, tipranavir, tenofovir, disoproxil fumarate, and zidovudine that are effective against diverse lethal infectious viral diseases [4]. Nevertheless, in addition to their therapeutic potential, these synthetic drug leads have been documented to induce numerous undesirable consequences [5]. In some instances, the antiviral agents may become ineffective in evolving virally resistant strains. With the steep rise in viral infections, the gap between production and demand for cost-effective, safe therapeutics increased drastically. Researchers began investigating transdisciplinary in silico, in vitro, and in vivo innovative ways to close the gap and help generate highly bioavailable, cost-effective, and target-specific synthetic and plant-derived antiviral medications within a set time frame.
During exploration, researchers found that the immune response modulating plant-derived metabolites can impede viral proliferation effectively without altering the host biological processes. With the advent of ethnopharmacology, researchers developed a vast number of phytotherapeutics from medicinal plants such as Achillea fragrantissima, Aegle marmelos, Aloe vera, Andrographis paniculata, Artocarpus integrifolia, Balanites aegyptiaca, Camellia sinensis, Capparis spinosa, Cassinexylocarpa, Cistus incanus, Dioscorea bulbifera, Echinacea augustofolia, Echinacea pallid, Echinacea purpurea, Glycyrrhiza uralensis, Lindera chunii, Nicotiana benthamiana, Phyllanthus emblica, Plantago major, Wistaria floribunda, and Xanthocerassorbifoli [6].Compared to all explored plants, the Salvadoraceae family grabbed the attention of ethnopharmacologists due to their unique bioactive metabolite’s availability and adaptability to arid environments. Among all the species of Salvadoraceae family Salvadora persica phytochemicals, salvadorine, salvadoricine, trimethylamine, benzyl isothiocyanate, and other flavonoids and steroids are extensively utilized in traditional and modern medicine to treat scabies, piles, leukoderma, asthma, cough, diabetes, and sexually transmitted bacterial and viral diseases because of their high bioavailability [7]. Although scientists have revealed the mechanism of action of Salvadora persica phytochemicals in communicable and noncommunicable diseases using cheminformatics, genomics, and proteomics approaches, knowledge of the mechanism of action on viral myocarditis is limited.
As a result, in the current study, we used various in silico screening strategies to identify powerful blood-brain barrier-crossing anti-Coxsackie B3 virus phytochemicals from Salvadora persica. We are certain that the collected results will be useful for undertaking in vitro and in vivo research to create preclinical and clinical data, which is required for the development of phytotherapeutics to treat CVB3-induced myocarditis.
MATERIALS AND METHODS
The in-silico screening study was conducted utilizing a Lenovo laptop equipped with an 11th-generation Intel(R) Core (TM) i3 processor and 8GB of RAM. The Swiss ADME web tool was utilized for ADME investigations. The PyRx and Biovia Discovery Studio software were utilized for the preparation of the target, ligand, followed by docking and visualization.
Preparation of ligands
The reference drugs Pleconaril, 2-Fucosyllactose, and Salvadora persica phytochemicals 3D structure were obtained from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) in SDF format. The list of all the compounds with their unique CID is as follows: Beta-sitosterol (CID:222284); Glucotropaeolin (CID:9573945); Trimethylamine (CID:1146); Methoxy benzoicacid (CID:11461); Sinigrin (CID:23671158); Myristicin (CID:4276); Myrcene (CID:31253); Longifolene (CID:289151); Benzyl isothiocyanate (CID:2346); Salvadourea (CID:18509381); Umbellulon (CID:91195); Benzaldehyde (CID:240); Beta-Elemene (CID:6918391); 3-Carene (CID:26049); Terpinolene (CID:11463); Humulene (CID:5281520); gamma-Muurolene (CID:12313020); alpha-Pinene (CID:6654); beta-Pinene (CID:14896); beta-Caryophyllene (CID:5281515); Camphene (CID:6616); Limonene (CID:22311); Myristic acid (CID:11005); Lauric acid (CID:3893); Palmitic acid (CID:985); Oleic acid (CID:445639); Liriodendrin (CID:21603207); Syringin (CID:5316860); 1-Triacontanol (CID:68972); 1-Octacosanol (CID:68406); Salvadoraside (CID:162343330); Salvadoside (CID:23664985); beta-sitosterol-3-O-beta-d-glucoside (CID:12309057); Liriodenine (CID:10144); 12-ethylidene-8,14-diazapentacyclo octadeca-2,4,6,17-tetraen-18-yl methanol (CID:14526586); Kaempferol (CID:5280863); Quercetin (CID:5280343); Normavacurine (CID:11969908); Rutin (CID:5280805); Quercitrin (CID:5280459) [8] and the standard antiviral drugs are Pleconaril (CID:1684) and 2-fucosyllactose (CID:170484) [9, 10]. The ligands were prepared by using PyRX software and used for further analysis.
ADME and drug-likeliness properties
Swiss ADME (http://www.swissadme.ch) is a low-cost online tool for assessing the pharmacokinetics, drug-likeness, medicinal chemistry, and compatibility of small compounds. ADME/T analysis is used in drug development to evaluate the absorption, distribution, metabolism, excretion, and toxicity of a specific ligand or chemical molecule. This technique is used to eliminate unwanted compounds that lack strong drug-like properties. The Swiss ADME online tool is used to analyze the pharmacological and physicochemical characteristics of chosen compounds or hits based on their canonical SMILES. Important criteria for pharmacological validation include molecular weight, hydrogen bond acceptor and donor, topological polar surface area (TPSA), LogS, blood-brain barrier penetration, and gastrointestinal absorption [11]. The efficient lead compounds that passed ADME are being investigated for in silico docking and interaction investigations.
Preparation of the target protein
The Coxsackie virus VP1 and VP2 capsid protein sequences, which play a critical role in adhesion to CAR receptors of the host system, were obtained from the National Center for Biotechnology Information (NCBI) with accession numbers 1JEW_1 and 1JEW_2, respectively. To build the tertiary structure of proteins essential for docking studies, the VP1 and VP2 protein sequences were submitted to the trRosetta web server [12]. To assess the stereo chemical stability of the proteins, the obtained structures were subjected to the PROCHECK web server, and loop modeling was performed to obtain the maximum residues in favorable regions by using the Galaxy loop interphase of the Galaxy web server and revalidated by the PROCHECK web server [13, 14]. The active site prediction web server tool (http://www.scfbio-iitd.res.in/dock/ActiveSite.jsp) was used to predict the active sites of the refined protein model. The selected target structures were used for in-silico analysis.
Molecular docking analysis
Docking assays were done using PyRx software to examine the binding affinity of ADME-passed ligands Umbellulon, Benzyl Isothiocyanate, Myrcene, Lauric Acid, and Methoxybenzoic Acid to VP1 and VP2. The ligands with the highest binding energy were then utilized to conduct molecular interaction analysis with Biovia Discovery Studio [15-17].
RESULTS AND DISCUSSION
ADME and drug-likeliness
Swiss-ADME predicts pharmacological efficacy by considering absorbability, bioavailability, site of action, metabolism, and excretion. Swiss-ADME tests were carried out on forty chosen phytochemicals from Salvadora persica. The notable features such as blood-brain barrier (BBB) permeation, gastrointestinal absorption, Lipinski's rule of five, including high oral bioavailability, polar surface area, and the presence of hydrogen bond acceptor and donor are considered for the selection of the best leads for docking given screening anti-Coxsackievirus therapeutics. The collected findings are presented in Table 1. The ADME cleared five blood-brain barrier crossing leads were processed for further study. During the study, it was also discovered that the BBB-permeable five molecules did not inhibit five major isoenzymes, CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4, which play important roles in the maintenance of cellular homeostasis and pharmacokinetics-related drug-drug interactions [18].
Table 1. ADME profile of phytochemicals
|
Molecules |
Molecular formula |
Molecula rweight |
Rotatablebonds |
H-acceptor |
H-donor |
TPSA |
ConsensusLogP |
Solubility |
GIabsorption |
BBB permeability |
Pgpsubstrate |
Inhibitors |
Lipinskiviolation |
Bioavailabilityscore |
|
Beta-sitosterol |
C29H50O |
414.7 |
6 |
1 |
1 |
20.2 |
8.02 |
Poorlysoluble |
Low |
No |
No |
No |
1 |
0.55 |
|
Glucotropaeolin |
C14H19NO9S2 |
409.43 |
7 |
10 |
5 |
199.79 |
-0.5 |
Yes |
Low |
No |
Yes |
No |
0 |
0.11 |
|
Trimethyl amine |
C3H9N |
59.11 |
0 |
1 |
0 |
3.24 |
0.34 |
Yes |
Low |
No |
No |
No |
0 |
0.55 |
|
Methoxy benzoic acid |
C8H8O3 |
152.15 |
2 |
3 |
1 |
46.53 |
1.49 |
Yes |
High |
Yes |
No |
No |
0 |
0.85 |
|
Sinigrin |
C10H17NO9S2 |
359.37 |
7 |
10 |
5 |
199.79 |
-1.27 |
Yes |
Low |
No |
Yes |
No |
0 |
0.11 |
|
Myristicin |
C11H12O3 |
192.21 |
3 |
3 |
0 |
27.69 |
2.49 |
Yes |
High |
Yes |
No |
Yes |
0 |
0.55 |
|
Myrcene |
C10H16 |
136.23 |
4 |
0 |
0 |
0 |
3.43 |
Yes |
Low |
Yes |
No |
No |
0 |
0.55 |
|
Longifolene |
C15H24 |
204.35 |
0 |
0 |
0 |
0 |
4.5 |
Yes |
Low |
No |
No |
Yes |
1 |
0.55 |
|
Benzyl isothiocyanate |
C8H7NS |
149.21 |
2 |
1 |
0 |
44.45 |
2.91 |
Yes |
High |
Yes |
No |
No |
0 |
0.55 |
|
Salvadourea |
C17H20N2O3 |
300.35 |
8 |
3 |
2 |
59.59 |
2.5 |
Yes |
High |
Yes |
No |
Yes |
0 |
0.55 |
|
Umbellulon |
C10H14O |
150.22 |
1 |
1 |
0 |
17.07 |
2.16 |
Yes |
High |
Yes |
No |
No |
0 |
0.55 |
|
Benzaldehyde |
C7H6O |
106.12 |
1 |
1 |
0 |
17.07 |
1.57 |
Yes |
High |
Yes |
No |
Yes |
0 |
0.55 |
|
Beta-Elemene |
C15H24 |
204.35 |
3 |
0 |
0 |
0 |
4.65 |
Yes |
Low |
No |
No |
Yes |
1 |
0.55 |
|
3-Carene |
C10H16 |
136.23 |
0 |
0 |
0 |
0 |
3.42 |
Yes |
Low |
Yes |
No |
Yes |
1 |
0.55 |
|
Tercinolene |
C10H16 |
136.23 |
0 |
0 |
0 |
0 |
3.4 |
Yes |
Low |
Yes |
No |
Yes |
0 |
0.55 |
|
Humulene |
C15H24 |
204.35 |
0 |
0 |
0 |
0 |
4.26 |
Yes |
Low |
No |
No |
Yes |
1 |
0.55 |
|
Gamma-Muurolene |
C15H24 |
204.35 |
1 |
0 |
0 |
0 |
4.17 |
Yes |
Low |
No |
No |
Yes |
1 |
0.55 |
|
Alpha-Pinene |
C10H16 |
136.23 |
0 |
0 |
0 |
0 |
3.44 |
Yes |
Low |
Yes |
No |
Yes |
1 |
0.55 |
|
Beta-Pinene |
C10H16 |
136.23 |
0 |
0 |
0 |
0 |
3.42 |
Yes |
Low |
Yes |
No |
Yes |
1 |
0.55 |
|
Beta-caryochyllene |
C15H24 |
204.35 |
0 |
0 |
0 |
0 |
4.24 |
Yes |
Low |
No |
No |
Yes |
1 |
0.55 |
|
Camphene |
C10H16 |
136.23 |
0 |
0 |
0 |
0 |
3.43 |
Yes |
Low |
Yes |
No |
Yes |
1 |
0.55 |
|
Limolene |
C10H16 |
136.23 |
1 |
0 |
0 |
0 |
3.35 |
Yes |
Low |
Yes |
No |
Yes |
0 |
0.55 |
|
Myristic acid |
C14H28O2 |
228.37 |
12 |
2 |
1 |
37.3 |
4.45 |
Yes |
High |
Yes |
No |
Yes |
0 |
0.85 |
|
Lauric acid |
C12H24O2 |
200.32 |
10 |
2 |
1 |
37.3 |
3.51 |
Yes |
High |
Yes |
No |
No |
0 |
0.85 |
|
Palmitic acid |
C16H32O2 |
256.42 |
14 |
2 |
1 |
37.3 |
5.2 |
Yes |
High |
Yes |
No |
Yes |
1 |
0.85 |
|
Oleic acid |
C18H34O2 |
282.46 |
15 |
2 |
1 |
37.3 |
5.65 |
Poorlysoluble |
High |
No |
No |
Yes |
1 |
0.85 |
|
Liriodenedrin |
C34H46O18 |
742.72 |
12 |
18 |
8 |
254.14 |
-1.85 |
Yes |
Low |
No |
Yes |
No |
3 |
0.17 |
|
Syringin |
C17H24O9 |
372.37 |
7 |
9 |
5 |
138.07 |
-0.48 |
Yes |
Low |
No |
No |
No |
0 |
0.55 |
|
1-Triaconcanol |
C30H62O |
438.83 |
28 |
1 |
1 |
20.23 |
10.92 |
Poorlysoluble |
Low |
No |
Yes |
No |
1 |
0.55 |
|
1-Octacosanol |
C28H58O |
410.8 |
26 |
1 |
1 |
20.23 |
10.14 |
Poorlysoluble |
Low |
No |
Yes |
No |
1 |
0.55 |
|
Salvadoraside |
C34H48O18 |
744.74 |
14 |
18 |
9 |
265.14 |
-2.38 |
Yes |
Low |
No |
Yes |
No |
3 |
0.17 |
|
Salvadoside |
C13H18O9S |
350.34 |
6 |
9 |
4 |
151.13 |
-0.7 |
Yes |
Low |
No |
No |
No |
0 |
0.11 |
|
Beta-sitisterol-3-O-Beta-d-glucoside |
C35H60O6 |
576.86 |
9 |
6 |
4 |
99.38 |
5.85 |
Poorlysoluble |
Low |
No |
No |
No |
2 |
0.55 |
|
Liriodenine |
C17H9NO3 |
275.26 |
0 |
4 |
0 |
48.42 |
2.88 |
Yes |
High |
Yes |
Yes |
Yes |
0 |
0.55 |
|
12-Ethylidene-8,14-diazapentacyclo octadeca-2,4,6,17-tetraen-18-yl methanol |
C19H22N2O |
294.39 |
1 |
2 |
2 |
35.5 |
2.41 |
Yes |
High |
Yes |
Yes |
Yes |
0 |
0.55 |
|
Kaempserol |
C12H24O2 |
200.32 |
10 |
2 |
1 |
37.3 |
3.51 |
Yes |
High |
Yes |
No |
No |
0 |
0.85 |
|
Quercetin |
C15H10O7 |
302.24 |
1 |
7 |
5 |
131.36 |
0.63 |
Yes |
High |
No |
No |
Yes |
0 |
0.55 |
|
Normavacurine |
C19H22N2O |
294.39 |
1 |
2 |
1 |
28.4 |
2.41 |
Yes |
High |
Yes |
Yes |
Yes |
0 |
0.55 |
|
Rutin |
C27H30016 |
610.5 |
6 |
16 |
10 |
266 |
-1.3 |
Soluble |
Low |
No |
Yes |
No |
3 |
0.17 |
|
Quercitrin |
C21H20O11 |
448.38 |
3 |
11 |
7 |
190.28 |
0.22 |
Yes |
Low |
No |
No |
No |
2 |
0.17 |
Structure validation
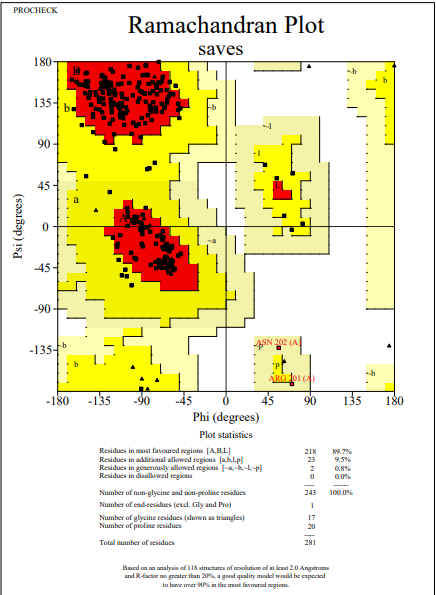
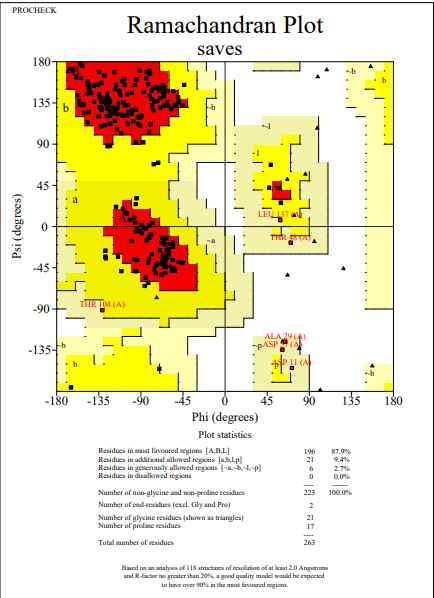
To check the quality of VP1 and VP2 protein tertiary structures obtained from trRosetta, the predicted model was initially subjected to the PROCHECK analysis. After analysis out of five models generated, the structure showing a percentage of favorable region of 89.3% for VP1 and 87.9% for VP2 was noticed and used for further analysis. To make the protein more stereochemically active, loop modeling was performed by using the Galaxy web server. Based on our insilico data analysis, it was noticed that the favorable region was 89.7% for VP1 and 87.9% for VP2 (Figures 1 and 2). This data indicates that after loop modeling, more stable structures were obtained, which are essential for in-silico binding affinity studies.
|
|
|
Figure 1. Ramachandran plot of VP2 after loop modelling |
|
Figure 5: Ramachandran plot of VP1 after loop modelling.
|
|
|
|
Figure 2. Ramachandran plot of VP2 after loop modelling |
Molecular docking and visualization
In virtual screening techniques, molecular docking is frequently employed to reduce massive libraries showing low binding affinity towards the receptor or target [19, 20]. This will also help drug discovery and development scientists conduct the in-silico, in-vitro, and in-vivo experiments cost-effectively in the stipulated time [21]. To explore the binding sites that exist on VP1 and VP2 essential for docking, the validated structures were subjected to active site prediction using the active site prediction web tool. The obtained active site coordinates, 31.62, -18.28, and 42.89 for VP1 and 17.64, 4.31, and 4.26 for VP2, were used to navigate the ligands toward binding pockets of targets to obtain better docking energies. The VP1 and VP2 targets were subjected to docking; during docking, water molecules and hetero atoms were eliminated from the structures, and polar hydrogen was added to facilitate the protein-ligand docking using the Biovia Discovery Studio program.
Docking of VP1
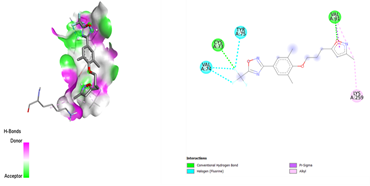
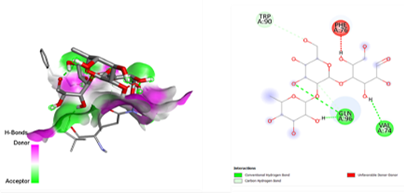
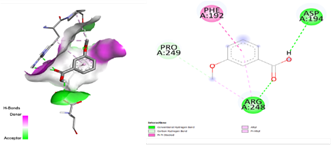
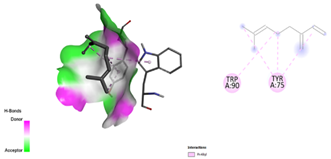
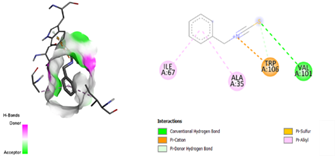
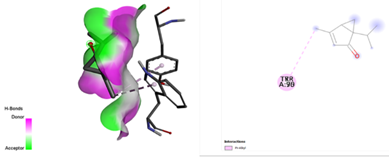
After docking the standard medication, pleconaril was discovered to have a significant binding affinity to the VP1 capsid protein, with a docking score of -6.1 kcal/mol. The contact involved typical hydrogen bonding with cysteine 73 and valine 91, indicating molecular specificity. Another typical medication, 2-fucosylactose, has a binding affinity with the VP1 capsid protein (docking score: -4.2 kcal/mol), forming two conventional hydrogen bonds with glutamine 96 and valine 74. The binding scores for the five phytochemicals ranged from -3.7 to -4.8 Kcal/mol. Notably, methoxybenzoic acid (CID 1461) had the closest binding affinity, with a score of -4.8 kcal/mol. Furthermore, the interaction between the ligand and target protein was determined by the formation of two hydrogen bonds involving essential amino acid residues, Arginine 248 and Aspartate 194. As a result, it was discovered that 3-methoxy benzoic acid had a nearly identical binding affinity for VP1 as the conventional medication, 2-fucosylactose. The acquired findings are reported in Tables 2 and 4.
Docking with VP2
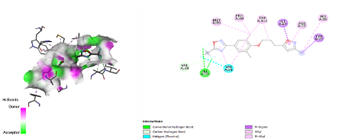
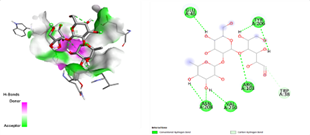
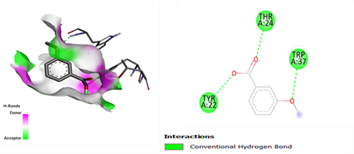
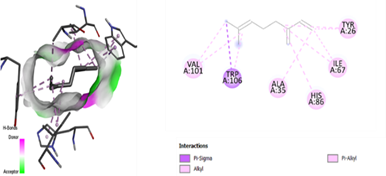
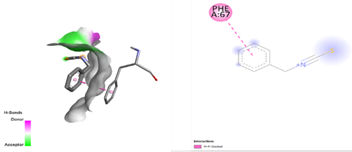
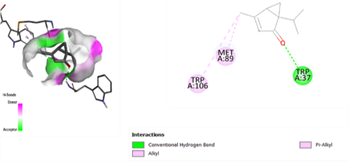
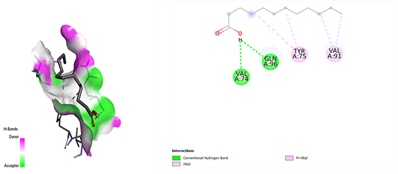
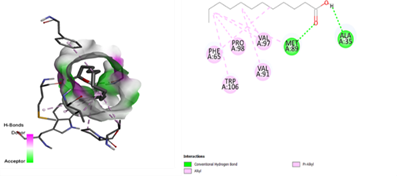
In the docking analysis, the standard drug pleconaril displayed a maximum binding affinity of -6.3 Kcal/mol with the VP2 capsid protein by forming the conventional hydrogen bond with isoléucine 39. In a parallel docking analysis of the second reference lead 2-fucosylactose, we also noticed that the lead molecule exhibited an almost similar binding affinity of -6.2 Kcal/mol towards the VP2 capsid protein. It was also observed that 2-fucosylactose formed five conventional hydrogen bonds with the VP2 capsid protein. This observation resonates with the overall trend, as it was noted that all five phytochemicals subjected to docking consistently yielded scores within the range of -5 to -5.8, emphasizing the uniformity in their binding affinities (Tables 3 and 4). Among the phytochemicals, it was observed that, except for myrecene (CID 31253), all the ligands bound to the target protein showcased hydrogen bond interactions, emphasizing the favorable binding characteristics of these compounds. Despite Umbellone (CID 91195) achieving a docking score of -5.8 against VP2 capsid proteins, which may seem less commendable compared to the standard drug, its distinct advantage makes it a notable candidate.
Table 2. Docking scores and amino acids involved in hydrogen bond formation between ligands and VP1
|
Compounds |
Binding Affinity (kcal/mol) |
Amino acid Residues involved in Conventional Hydrogen Bond formation |
|
Pleconaril |
-6.1 |
CYS-73, VAL-91 |
|
2-Fucosyl lactose |
-4.2 |
GLN-96, VAL-74 |
|
Methoxy benzoic acid |
-4.8 |
ASP-194, ARG-248 |
|
Myrecene |
-3.7 |
Nil |
|
Benzyl isothiocyanate |
-4.5 |
Nil |
|
Umbellulon |
-4 |
Nil |
|
Lauric acid |
-4 |
VAL-74, GLN-96 |
Table 3. Docking scores and amino acids involved in hydrogen bond formation between ligands and VP2
|
Compounds |
Binding Affinity (kcal/mol) |
Amino acid Residues involved in Conventional Hydrogen Bond formation |
|
Pleconaril |
-6.3 |
ILE 39 |
|
2-Fucosyl lactose |
-6.2 |
GLU-46, ARG-103, TYR-206, ASN-208 and VAL-210 |
|
Methoxy benzoic acid |
-5.6 |
TYR-22, THR-24, TRP-37 |
|
Myrecene |
-5.1 |
Nil |
|
Benzyl isothiocyanate |
-5.1 |
VAL-101 |
|
Umbellulon |
-5.8 |
TRP-37 |
|
Lauric acid |
-5 |
ALA-35 and MET-89 |
Table 4. VP1 and VP2 Amino Acids 2D interaction with the ligands
|
Ligand Name |
VP1 Amino Acids 2D interaction with the ligands. |
VP2 Amino Acids 2D interaction with the ligands. |
|
Pleconaril |
|
|
|
2-Fucosyl lactose |
|
|
|
Methoxy benzoic acid |
|
|
|
Myrecene |
|
|
|
Benzyl isothiocyanate |
|
|
|
Umbellulon |
|
|
|
Lauric acid |
|
|
CONCLUSION
In investigating Coxsackie virus B3-induced myocarditis, our study focused on targeting the viral capsid proteins V1 and VP2, which play a pivotal role in facilitating viral entry into myocytes. Recognizing Salvadora persica’s reported antiviral properties to inhibit the Herpes simplex virus, we conducted an in-silico phytochemical screening analysis to identify potential therapeutic leads essential for the management of viral myocarditis. Based on our studies, we observed that BBB permeable methoxy benzoic acid and umbellone are the potential phytochemicals to be explored in-silico, in-vitro, and in-vivo data generation, essential for the management of myocarditis.
Acknowledgments: The authors would like to express gratitude to the Indian Academy Institutions management for providing the infrastructural facilities to carry out this work.
Conflict of interest: None
Financial support: None
Ethics statement: None