International Journal of Pharmaceutical and Phytopharmacological Research
ISSN (Print): 2250-1029
ISSN (Online): 2249-6084
|
Template-based In-Silico Studies on Lipopolysaccharide-Binding Protein
Vamshi Venkat M, Vaishnavi Mallojala, Shravan Kumar Gunda*, Mahmood Shaik |
|
Bioinformatics Division, PGRRCDE, Osmania University, Hyderabad, India |
|
ABSTRACT |
|
Lipopolysaccharide-binding protein is responsible for the natural defense reaction. LBP binds to lipid A moiety of the bacterial lipopolysaccharide (LPS), a glycolipid present in the outer membrane of all Gram-negative microorganisms. Lipopolysaccharide acts as a binding enhancer to CD14, and on cellular surfaces, CD14 behaves as a dissolvable protein and also as an acceptor of LPS in the linked form of phosphatidylinositol. In the present study, we generated a precise 3-Dimensional model of LBP using MODELLER 9.21 and validated its structure using Procheck software. The optimum number of amino acids were found in the core region of the modeled protein. We interpreted the action of natural compounds docking against the modeled LBP protein. Three compounds (Ginkgetin, Linderatone, and Erystagallin A) showed lower binding affinity values towards LBP compared to Diclofenac, Celecoxib, and Indomethacin. Ginkgetin exhibited the lowest binding energy of -11.77 Kcal/mol by interacting with His294 and Lys467. Binding energies of all the twenty compounds manifested exceptionally than the standard drugs for the modeled LBP protein. These computational studies can help discover novel drug candidates. |
|
Key Words: LBP, Homology Modelling, Molecular Docking, Natural compounds, CD14. |
INTRODUCTION
The first 60-kDa glycoprotein was isolated from rabbit serum 25 years ago, which is the lipopolysaccharide-binding protein that was further considered as an active protein that can control LPS properties biochemically and organically. As the LPS-binding protein attaches to lipopolysaccharide, LPS-mediated effects are observed strongly on immune cells, which is enhanced by the LPS-binding protein. LPS/LBP acts as an acute-phase protein and provides a crucial aspect in the intrinsic reaction to bacterial tests. It is a potential biomarker in examining the progression of serious pyemia, irresistible endocarditis, and cardiovascular disease.[1, 2] Receptors of macrophages, monocytes, granulocytes and dendritic cells that exhibit multiple functions during immune response identify the specific attacking microorganism inside the host. [3] Various microorganisms possess the primary association with the host upper respiratory tract, where they can act as potential pathogens.[4]
Lipopolysaccharide (LPS) plays a significant role in the pathological process of serious pyemia and instant septic poisoning in gram-negative microorganisms.[5] Certain macrophages and monocytes are stimulated in-order to release cytokines [6]. Plasma lipopolysaccharide-binding protein and CD14 membrane are the two vital proteins that control the pathway of circulatory monocytes. Neutralization of anti-LBP and anti-CD14 antibodies are known to exhibit the repression of LPS-induced proinflammatory responses in vivo and in vitro.[7]
The lipoproteins of treponemes have been appeared to be strong destructive components; i.e., the lipoproteins of T. pallidum activate NF-B translocation in monocytes and those of T. denticola enact in large-scale phages.[8] The incitement of the Kupffer cell by the endotoxin triggers irritation, for example, tumor necrosis factor (TNF), is to a high-degree helpful model in which biochemical changes can be identified with the seriousness of obsessive liver damage[9]. Some of the studies suggest that LBP has a role in Escherichia coli peritonitis.[10] CD14 gets attached to LBP of the Gram-negative microorganism, and it transfers such components to Toll-like receptors (TLR), which further triggers the cell activation. Accessory receptors regulate this mechanism in the lungs including the acute phase protein (LBP) and some surfactant proteins where CD14 regulates LPS-induced activation of the cells with very stunted expression of CD14, which is either beneficial to host in the inflammatory immune response to eradicate microbes or causes excessive inflammation or overstimulation.[11]
LBP can likewise restrain reactions to LPS by complex systems that rely on LBP concentration and the cell condition. When directed intra-peritoneal, LBP can protect host from the harmful effects of Gram-negative bacteria. [12] When LBP levels increase both in the plasma and extravascular fluids in the middle of Gram-negative sepsis, it might help control reactions to LPS apparently because of their immunosuppressive impacts. The dual activities of sCD14 (soluble CD14) or LBP may give a method of reasoning for their potential advantages as therapeutics, which must be researched.[13] LBP plays a role in aspiratory recognition of Gram-negative microbes and the natural invulnerable reaction to pneumonia. The decreased survival in LBP mice is related to impeded leeway of microorganisms from the lungs and would prompt quantifiable misperceptions in the pneumonic reactions in a TLR4 subordinate way.[14]
In the current study, accurate in the silico-structure analysis was performed due to the nonappearance of crystal structure for Lipopolysaccharide-binding protein (Accession number P18428). Modeler 9.21 was used as a tool for the construction of the predicted protein model validation was performed with the use of PROCHECK and to study the binding affinities of the protein-molecular along with ligand docking studies were performed by using the specific tool autodock 4.2.
METHODOLOGY
Sequence alignment and structure prediction
The sequence of amino acids of the Lipopolysaccharide-binding protein (Accession number P18428) from the species Homo sapiens was retrieved from the UniProtKB database (http://www.uniprot.org/)[15]. A BLAST (Basic Local Alignment Search Tool) search was performed to select the template and resulted with the best match Crystal structure of Chain A, Lipopolysaccharide-binding Protein) (PDB ID: 4M4D_A)[16] with 69% similarity having a resolution of 2.91 Å making it an outstanding template. A 3-D predicted model was constructed using Modeller 9.21. The respective templates were retrieved from protein databases like PDB[17, 18]. It is essential to consider the template’s resolution and sequence identity in opting for the template. The resulting model would be sufficiently good to allow structural and functional research only when both parameters are high. Using PROCHECK a model with final validation was achieved for the Ramachandran plot. Further, the calculation of RMSD was performed by superimposing (4M4D) over the generated model to evaluate the reliability and accuracy of the model.
Figure 1: Query sequence alignment of (LBP) and template sequence of (4M4D)
To achieve satisfactory results in the model, we further used MODELLER 9.21[19], which is an automated approach to homology modeling with spatial restraints. ClustalX[20] and Clustalw2[21] then opted for sequence alignments using the protein and template sequences, which are shown in figure 1. Construction of Homology models for the chosen protein was performed using Modeller 9.21. Later on the modification of the alignment input file, we generated 20 different models and chose the best model among them, which was determined by the lowest modeler objective function value. Models with Stereochemical quality were evaluated by using specific program software like PROCHECK[22], where the model is used in studying the structural and functional aspects. Ramachandran plot explains the listing of all the residues, which facilitates the psi/phi angles calculation and conformation of the backbone of the models.
Docking methodology
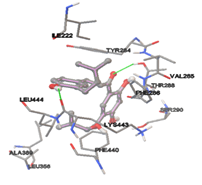
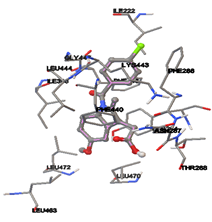
By using Tripo’s Sybyl6.7 the active site was predicted. It showed three active site pockets. The amino acids presented in pocket one are Ile222, Ala226, Asn27, Lys248, Gly249, Gly29, Ala264, Leu30, Ile281, Ser282, Asp283, Tyr284, Val285, Leu391, Ile398, Lys443, Leu444, Glu446, Gly447, and Phe448.
Twenty natural compounds were downloaded from NCBI and saved into .mol2 format. Molecular Docking study was performed to all the natural ligands separately by using the AutoDock4.2 [23] program. Upon using the Lamarckian genetic algorithm, the empirical free energy function is implemented. Initially, the modeled Lipopolysaccharide-binding protein was loaded, and besides, hydrogens were added, which was later saved in the format of PDBQT. Further, the initial loading of the ligand with the proper setting of conformations was done and saved in PDBQT format. Specific grid parameters were calculated in the auto grid. For every docking, a grid point with spacing 0.375 Å was applied, then a precise grid map considering 60×60×60 points, prediction of active site was identified by using sybyl6.7 a biopolymer module. X, Y, Z coordinated were opted by the presence of various amino acids that were identified in the active site region. Later, the Auto dock was run with default set parameters.
RESULTS AND DISCUSSION:
Homology modeling with model evaluation:
The current study reported the protein (PDB ID: 4M4D_A) having a high degree of homology with P18428 protein, which was used as a template, with the excellent atomic resolution concerning its crystal structure. The query sequence of Chain A, Lipopolysaccharide-binding Protein from Homo sapiens having 481 amino acid residues, was retrieved from the UniProt protein sequence database with Accession No. P18428. PDB Id 4M4D_A was identified and selected as a template using BLAST having 69% identity and 83% positives.
Upon using modeler 9.21, the structure was generated. It was validated with the help of its protein structure and PROCHECK where the generated model exhibited 91.3% of amino acid residues with 387 amino acids in the core region, 8.5% of amino acid residues with 36 amino acids in the additionally allowed region where 0.2% of amino acid residues with 1 amino acid in the moderately allowed region and there were no amino acids in the part of disallowed.
The template PDB shows amino acid residues of 81.7% with 629 amino acids in the core region, 17% with 131 in the additionally allowed region, the other amino acid residues of 1.3% with 10 amino acids in the moderately allowed region and there are none in the disallowed region. The cartoon model is shown in fig.2 and the Ramachandran plot of modeled protein is demonstrated in fig.3. Calculation of the root mean square deviation i.e RMSD was done for the generated model and template by using SPDB Viewer. The two models were loaded and are superimposed together using carbon alpha and calculated RMSD. The superimposed model is shown in figure 4. RMSD was observed with 0.46 Å, which indicates the generated model exhibited a similar function as a template.
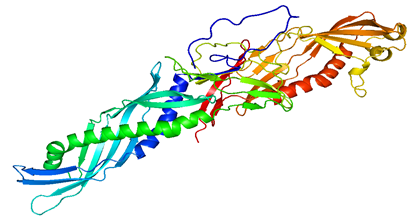
Figure 2: Cartoon model of modeled LBP protein
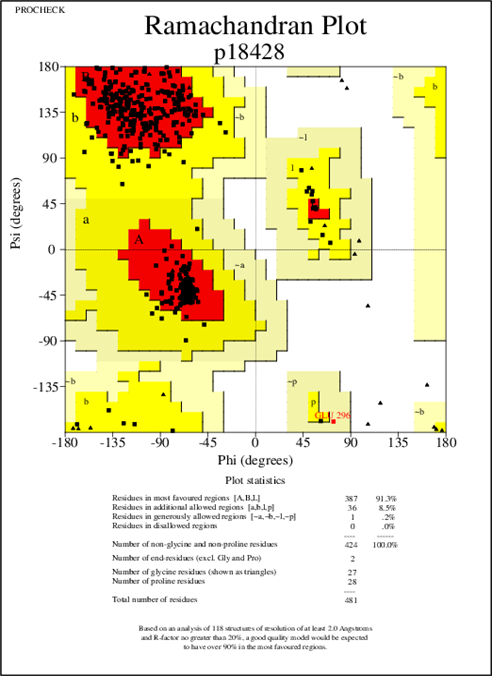
Figure 3: Ramachandan plot of the modeled LBP Protein
Figure 4: Superimposed model of modeled LBP (query) and 4M4D (template)
Molecular docking results:
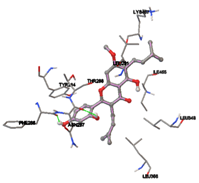
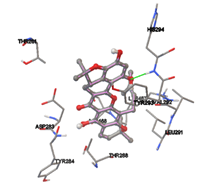
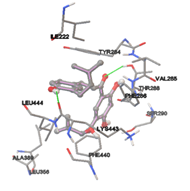
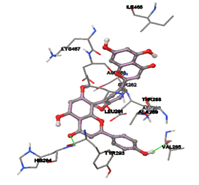
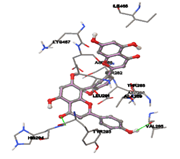
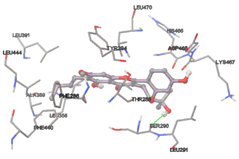
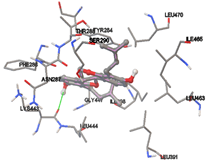
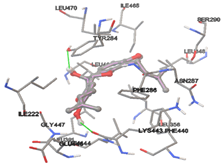
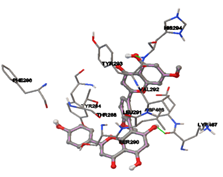
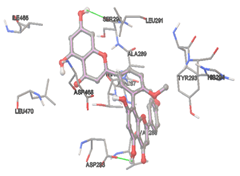
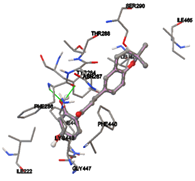
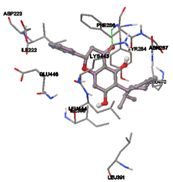
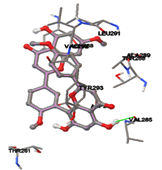
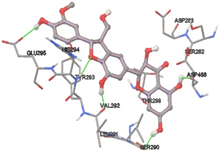
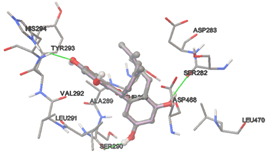
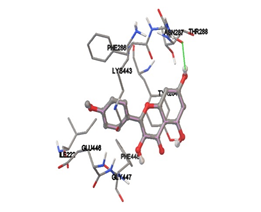
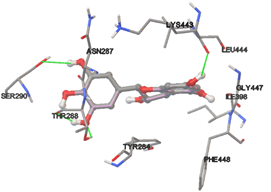
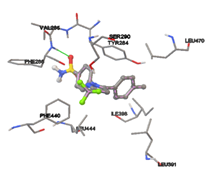
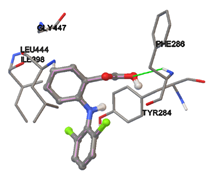
One of the efficient and extensively used methods for the calculation of protein-molecule (ligand) interactions is molecular docking. It is one of the best ways for the prediction of the relationship between proteins and ligands. In the present study, the native plant flavonoid, terpenoids, etc., which are also called secondary metabolites (ligands), have been observed as Lipopolysaccharide-binding protein inhibitors. AutoDock4.2 uses free energy binding calculation to allocate the best binding conformation. Twenty potent molecules were allowed for docking and also used three already existing drugs as standards for docking study against the modeled protein. Docking study has shown the H-bonding of Ginkgetin with His294 and Lys467 residues of modeled protein with a docking score value of -11.77 Kcal/mol. Compound Linderatone exhibited binding energy of -11.02 Kcal/mol with interacting Phe286, and Erystagallin A exhibited binding energy of -10.31 Kcal/mol with interacting Tyr284 and Lys443. Three compounds exhibited binding energy of -10.00 Kcal/mol. Ten compounds with binding energy values ranging from 8.00 to 10.00 Kcal/mol are exhibited.
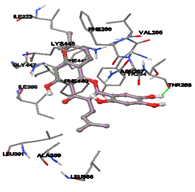
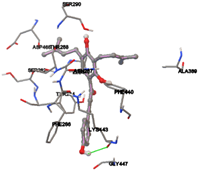
The compound Silicristin showed more interactions of Asp468, Ser90, Val292, His294, and Glu295 with the binding energy of -7.70 Kcal/mol. Epigallocatechin exhibited binding energy of -7.30 Kcal/mol with the interactions of Thr288, Ser290, Leu444, and Thr288. All the compound interactions and binding energies are demonstrated in Table 1 and figure 5.
Table 1: binding energies and their interacting amino acid residues of all the twenty docked compounds.
|
S.No |
Compound Name |
Interacting amino acids |
Binding energy ΔG (Kcal/Mol) |
Dissociation Constant (uM) |
|
1 |
Artocarpin |
Asn287, Thr288 |
-7.44 |
3.55uM |
|
2 |
Artonin |
His294 |
-7.83 |
1.81uM |
|
3 |
Artonin E |
Thr288 |
-9.90 |
55.39nM |
|
4 |
Cudraflavone |
Thr288, Ser90 |
-8.99 |
257.27nM |
|
5 |
Lonchocarpol A |
His294, Asp468 |
-6.72 |
11.92uM |
|
6 |
Bartericin A |
Thr288, Lys443 |
-8.74 |
390.8nM |
|
7 |
Bilobetin |
His294, Val285 |
-8.56 |
535.11nM |
|
8 |
Chaplashin |
Ser290 |
-8.29 |
833.17nM |
|
9 |
Erybraedin |
Lys443 |
-9.43 |
121.55nM |
|
10 |
Erystagallin A |
Tyr284, Lys443 |
-10.31 |
27.82nM |
|
11 |
Ginkgetin |
His294, Lys467 |
-11.77 |
2.38nM |
|
12 |
Isoginkgetin |
Asp283 |
-8.69 |
430.31nM |
|
13 |
Kanzonol B |
Phe286, Asn287 |
-8.09 |
255.09nm |
|
14 |
Linderatone |
Phe286 |
-11.02 |
8.31nM |
|
15 |
Medicagenin |
Lys443 |
-8.71 |
415.51nM |
|
16 |
Sciadopitysin |
Val285 |
-7.12 |
6.04uM |
|
17 |
Silicristin |
Asp468, Ser90, Val292, His294, Glu295 |
-7.70 |
2.27uM |
|
18 |
Vogelin C |
His294, Ser290, Ser282 |
-6.93 |
8.33uM |
|
19 |
Kaemferide |
Asn287 |
7.61 |
2.65uM |
|
20 |
Epigallocatechin |
Thr288, Ser290, Leu444, Thr288 |
-7.34 |
4.16uM |
|
21 |
Celecoxib |
- |
-7.77 |
2.03uM |
|
22 |
Diclofenac |
Phe286 |
-7.25 |
4.85uM |
|
23 |
Indomethacin |
- |
-9.82 |
62.89nM |
|
1 |
2 |
|
3 |
4 |
|
5 |
6 |
|
7 |
8 |
|
9 |
10 |
|
11 |
12 |
|
13 |
14 |
|
15 |
16 |
|
17 |
18 |
|
19 |
20 |
|
21 |
22 |
|
23 |
|
Figure 5: Docking of twenty natural compounds and standard drugs against modeled LBP protein.
CONCLUSION
The 3-Dimensional model of the LBP from Homo sapiens was developed using the molecular modeling method. To predict the model, Chain A, Lipopolysaccharide-binding Protein, was used as a template. The expected model showed 91.3% of the maximum number of amino acid residues in the highly favored region. The active site amino acid residues of the LBP model were identified. Molecular docking studies were also achieved to the modeled protein by taking all twenty natural compounds. Every compound exhibited the lowest binding energies and interactions than already existing drugs, which may lead to potent inhibitors for LBP.
REFERENCES