International Journal of Pharmaceutical and Phytopharmacological Research
ISSN (Print): 2250-1029
ISSN (Online): 2249-6084
|
In-silico Molecular Docking, ADME and Drug Likeness Predictions of Some α-alkylidene-β-ethoxycarbonyl Cyclopentanones
Feten Beji1,2, Reem al Harbi1, Arshi Naqvi1,3* |
|
1 Department of Chemistry, Taibah University, 30002 Al-Madinah Al-Munawarah, Saudi Arabia. 2Laboratory of Heteroatom Organic Chemistry, University of Carthage, Faculty of Sciences of Bizerte, 7021-Jarzouna, Tunisia. 3BioDiscovery-Solutions for future, Plot No 29, FF2, 2nd street, Pearl Astragal Apartment, Perumbakkam, off Medavakkam, Solinganallur Main Road, Chennai (Tamil Nadu)-600100, India. |
ABSTRACT
Computer-aided drug design (CADD) conception in the present-day scenario has transfigured the total drug discovery and development program. The pharmacodynamic and pharmacokinetic aspects of a drug candidate play a key role in deciding its fate as a therapeutic agent. The pharmacokinetic fortune of any drug candidate is determined by various properties like its absorption, distribution, metabolism, and elimination (ADME). In this report, an in-silico examination of α-alkylidene-β-ethoxycarbonyl cyclopentanones (1-7) was persuaded based on various physico-chemical parameters to predict their drug-likeness, pharmacokinetic and docking profile utilizing different computational tools.
Key Words: Sarkomycin, in-silico molecular docking, CADD, Drug likeness, ADME.
INTRODUCTION
Pharmaceutical consultation is an integral part of the retail sale of drugs to the public [1]. Pharmaceutical services are a critical component of primary health care [2]. The liposomal formulations are targeted to deliver the important drug combinations to the body [3]. Quality of life is the general well-being of individuals and communities [4]. In search of new drugs, (R)-Sarkomycin (1) was isolated for the first time in the year 1953 by Umezawa from the Streptomyces erythrochromogenes, a soil microorganism [5]. Its structure was established in 1955 as 2-methylene-3-oxocyclopentanecarboxylic acid [6]. This cyclopentenone had swiftly captured relevance for its antibiotic as well as strong anti-tumor activity [7-9]. It is well known and explored for its strong inhibitory effect on several human tumors and carcinoma cell lines [10-13]. Since it is chemically unstable [14], various stable sarkomycin esters (2) and the so-called cyclosarkomycin, which is a cyclic lactone (3), have been developed [14-19]. (Figure 1)
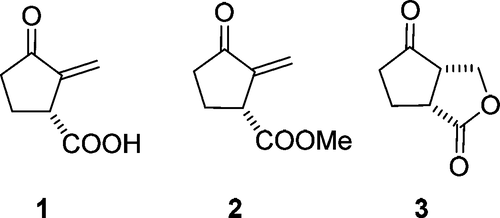
Figure 1: Some sarkomycin structures.
Mere synthesis of a chemical compound does not automatically make it an appealing candidate for drug development but certain attributes differentiate a drug from a synthesized compound. Several pharmacokinetic and pharmacodynamic properties are involved in deciding the drug candidature of a synthesized compound. In this regards, Computer-aided drug designing (CADD) or in silico notion is considered as it assists and expedites hit identification, facilitate assortment of hit-to-lead, sieve out a huge library of compounds into mini clusters of predicted bio-active compounds which in turn optimizes the ADME (absorption, distribution, metabolism, and excretion) profile leading avoiding issues related to safety and failure. Thus, assessment of ADME traits at the premature stages of drug discovery not only saves time but also money.
Phosphoinositide 3-kinases (PI3Ks), a member of lipid kinases family is interpreted to perform a pivotal role in nurturing survival and cellular growth of various sorts of cancer. In cancer cells, these PI3Ks are the essential regulators of apoptosis. The phosphorylation of phosphatidylinositol of the inositol ring at the 3′-OH position is catalyzed by the PI3Ks, thus obtaining secondary messenger lipid named phosphatidylinositol-3,4,5-trisphosphate. This trisphosphate triggers the serine/threonine kinase AKT which in return adjusts various signaling pathways thus, regulating apoptosis, survival of cells, proliferation of cells, and motility [20, 21]. PI3KCA gene encoding p110a is the most usually mutated gene in human tumors, thus instilling to be the most salient focus for tumor therapy [22-24]. Molecular docking studies were conducted to get an additional impression of the binding style of the synthesized compounds with PI3K p110α domain (PDB ID: 2RD0).
Our major intention is to evaluate our previously reported sarkomycin ester derivatives [25] for their pharmacokinetics, drug-likeness, and docking scores based on various physico-chemical parameters via in-silico computational tools.
EXPERIMENTAL
Ligand identification
The explored sarkomycin ester derivatives have been synthesized and characterized previously [25]. They are utilized as drug molecules for the present study.
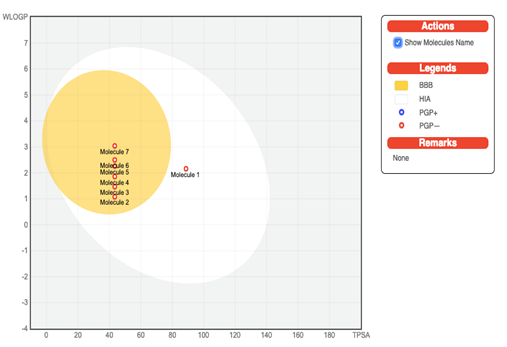
Figure 1: Structures of the selected compounds 1-7 for in-silico studies.
In-silico predictive studies
MolSoft renders several software and service tools for grasping the spatial framework of drug candidates, their biological substrates, facilitate understanding their interactions with each other
at the atomic level by applying a set of rules and algorithms to peculiar biomedical issues. Drug-likeness is one of the qualitative ideas employed for predicting drug-like property. It is designated as an intricate balance of diverse molecular and structural features which plays a pivotal task in establishing whether the specific drug candidate is alike the known drugs or not.
The targeted molecules 1-7 were appraised for predicting the drug-likeness based on 5 separate filters namely Egan [26], Ghose [27], Muegge [28], Veber [29] and Lipinski [30] rules accompanying bioavailability and drug-likeness scores using the Molsoft software and SwissADME program. Besides, SwissADME was also utilized to predict the physico-chemical, pharmacokinetic, and ADME properties.
In-silico molecular docking approach was used to do drug designing based on structure designing to obtain the more powerful inhibitors of PI3K from the synthesized drug candidates. 3D atomic coordinates of the ligands were drawn utilizing the ACD/Labs-Chemsketch program. The PDB ID, 2RD0 was considered to be the most relevant pure 3D crystal structure of p110α subunit of PI3K which is obtained from the protein data bank (PDB) (Source: www.rcsb.org/pdb/). Minimization of the energy of the molecules was done using the Dundee PRODRG2 server [31]. Autodock4 from “Auto-Dock Tools (ADT, 1.5.6)” which is a graphical user interface program was utilized for the docking studies [32]. The Lamarckian Genetic Algorithm (LGA) was chosen over the Monte Carlo method (present in previous versions of AutoDock). Kollman united-atom charges were allocated along with the incorporation of only polar hydrogens to the protein receptor. Gasteiger charge was allotted to and torsions were set. The grids were constructed and tuned in X, Y, Z-axis so that the whole active site of the targeted protein is covered. The docking statements were positioned to the software's default values and a standard etiquette was preserved and escorted throughout the docking evaluations. The results were annotated based on .pdf file created by the software by rating the various ligands concerning the foreseen binding energy. A cluster investigation that is based on values of root means square deviation (RMSD) was administered. The cluster acquiring the lowest energy value was reviewed as the most authentic solution. 3D visualization of the ligand-protein association was done by employing UCSF Chimera 1.11.2 and mcule, a web interface which utilizes the WebGL/Javascript based molecule viewer of GLmol.
RESULT AND DISCUSSION
The research work outlined here was conducted with the purpose to forecast the physicochemical attributes, drug-likeness, pharmacokinetic/ADME properties, and molecular docking simulations of our previously reported sarkomycin ester derivatives 1-7 using in-silico tools.
Several physicochemical traits like no. of rotatable bonds, count of specific atom types, molecular refractivity, lipophilicity, and water solubility were listed. TPSA (Topological Polar Surface Area) is a basic physiochemical variable gauged for assessing drug transport features. Calculations were undertaken to predict % Absorption for all the targeted compounds by the reported formula (%ABS = 109-(0.345 X TPSA)) [33]. All the tested compounds demonstrated excellent in-silico % absorption with the highest being 94.04%. These physicochemical properties are given in Table 1.
Table 1: Physicochemical properties of the selected compounds 1-7.
|
Comp. No. |
Fraction Csp3a |
No. of rotatable bonds |
HBAb |
HBDc |
iLogPd |
M.Re |
Log Sf |
TPSAg |
In-silico % absorption |
|
1 |
0.83 |
8 |
6 |
0 |
2.47 |
70 |
S |
88.71 |
78.40 |
|
2 |
0.56 |
3 |
3 |
0 |
1.98 |
44.27 |
S |
43.37 |
94.04 |
|
3 |
0.6 |
3 |
3 |
0 |
2.19 |
49.08 |
S |
43.37 |
94.04 |
|
4 |
0.64 |
4 |
3 |
0 |
2.05 |
53.89 |
S |
43.37 |
94.04 |
|
5 |
0.67 |
5 |
3 |
0 |
2.68 |
58.69 |
S |
43.37 |
94.04 |
|
6 |
0.69 |
5 |
3 |
0 |
2.82 |
63.5 |
S |
43.37 |
94.04 |
|
7 |
0.71 |
7 |
3 |
0 |
3.16 |
68.31 |
S |
43.37 |
94.04 |
aThe ratio of sp3 hybridized carbons over the total carbon count of the molecule; bnumber of hydrogen bond acceptors; cnumber of hydrogen bond donors; d lipophilicity; eMolar refractivity, fWater solubility (SILICOS-IT [S=Soluble]); gtopological polar surface area (Å2).
Forecast for the pharmacokinetic/ADME properties of the screened compounds 1-7 is given in Table 2. All of the tested compounds displayed high gastrointestinal (GI) absorption and are P-gp (p-glycoprotein) non-inhibitors. All the tested molecules were able to pass the blood-brain barrier (BBB) except compound 1. The predictions for the passive BBB permeation, HIA (human gastrointestinal absorption), and P-gp substrates are displayed together in the BOILED-Egg diagram as shown in Figure 2. None of the tested compounds inhibit the Cytochrome P450 isomers except compound 7 which inhibits CYP1A2 and CYP2C19. The compounds emerged to be low on skin permeability.
Table 2. Pharmacokinetic/ADME properties of the selected compounds 1-7.
|
Comp. No |
Pharmacokinetic/ADME properties |
||||||||
|
GI absa |
BBB permeantb |
P-gp substratec |
CYP1A2 inhibitord |
CYP2C19 inhibitore |
CYP2C9 inhibitorf |
CYP2D6 inhibitorg |
CYP3A4 inhibitorh |
Log Kpi |
|
|
1 |
High |
No |
No |
No |
No |
No |
No |
No |
-7.82 |
|
2 |
High |
Yes |
No |
No |
No |
No |
No |
No |
-6.69 |
|
3 |
High |
Yes |
No |
No |
No |
No |
No |
No |
-6.61 |
|
4 |
High |
Yes |
No |
No |
No |
No |
No |
No |
-6.39 |
|
5 |
High |
Yes |
No |
No |
No |
No |
No |
No |
-6.09 |
|
6 |
High |
Yes |
No |
No |
No |
No |
No |
No |
-6 |
|
7 |
High |
Yes |
No |
Yes |
Yes |
No |
No |
No |
-5.5 |
aGastro Intestinal absorption, bBlood Brain Barrier permeant, cP-glycoprotein substrate, d CYP1A2: Cytochrome P450 family 1 subfamily A member 2 (PDB:2HI4), e CYP2C19: Cytochrome P450 family 2 subfamily C member 19 (PDB:4GQS), fCYP2C9: Cytochrome P450 family 2 subfamily C member 9 (PDB:1OG2), g CYP2D6: Cytochrome P450 family 2 subfamily D member 6 (PDB:5TFT), h CYP3A4: Cytochrome P450 family 3 subfamily A member 4 (PDB:4K9T), iSkin permeation in cm/s.
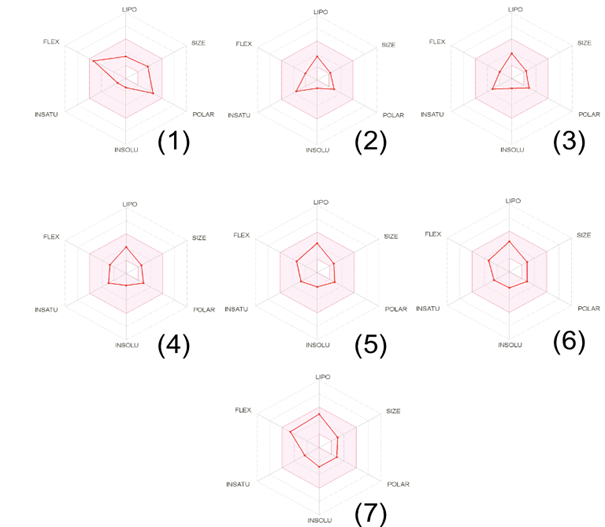
Figure 2: BOILED-Egg diagram of the selected compounds 1-7.
Drug likeness is examined as an integral element that provides the base for the molecules to be a powerful drug aspirant. Various rules viz. Lipinski, Ghose, Veber, Egan, and Muegge were considered to gauge drug-likeness of the candidate compounds (1-7) to find out whether they can be bioactive drug candidates according to some acute criterion like molecular weight, LogP, number of hydrogen bond acceptors and donors. The number of violations to the above-disclosed rules along with bioavailability and drug-likeness scores is given in Table 3. None of the compounds 1-7 violated any rule except for compounds 2,3 and 4 which are discarded with one violation according to Muegge rule. All the screened compounds disclosed bioavailability score around 0.55 and exhibited moderate to good drug-likeness scores ranged from -1.13 to
-0.30. The bioavailability radar of the compounds 1-7 is displayed in figure 3. The prime in-silico conformation was corroborated utilizing molecular docking screenings of the compounds 1-7 on p110α subunit of PI3K (PDB ID: 2RD0) as the target protein. To entertain this objective AutoDock 4.0, a Lamarckian genetic algorithm docking program was involved. Docking of the ligand 6 is demonstrated in Figure 4 and the docking scores are reported in Table 3. Docking scores range from -5.1 to -6.2 where the maximum docking score was obtained by compound 6 i.e -6.2.
Table 3. Drug likeness predictions and docking scores of the selected compounds 1-7.
|
Comp. No. |
Lipinski violations |
Ghose violations |
Veber violations |
Egan violations |
Muegge violations |
Bioavailability Score |
Drug Likeness score |
Docking score |
|
1 |
0 |
0 |
0 |
0 |
0 |
0.55 |
-0.72 |
-5.5 |
|
2 |
0 |
0 |
0 |
0 |
1 |
0.55 |
-1.13 |
-5.1 |
|
3 |
0 |
0 |
0 |
0 |
1 |
0.55 |
-0.77 |
-5.6 |
|
4 |
0 |
0 |
0 |
0 |
1 |
0.55 |
-0.72 |
-5.4 |
|
5 |
0 |
0 |
0 |
0 |
0 |
0.55 |
-0.68 |
-5.6 |
|
6 |
0 |
0 |
0 |
0 |
0 |
0.55 |
-0.30 |
-6.2 |
|
7 |
0 |
0 |
0 |
0 |
0 |
0.55 |
-0.73 |
-5.8 |
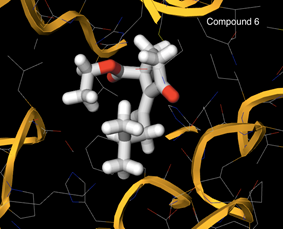
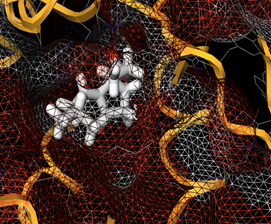
Figure 3: Bioavailability Radar of the tested compounds (1-7) The pink area represents the optimal range for each property (lipophilicity: XLOGP3 between − 0.7 and + 5.0, size: MW between 150 and 500 g/mol, polarity: TPSA between 20 and 130 (Å2)., solubility: log S not higher than 6, saturation: the fraction of carbons in the sp3 hybridization not less than 0.25, and flexibility: no more than 9 rotatable bonds. In this example, the compound is predicted not orally bioavailable, because too flexible and too polar.
Figure 4: Docking of compound 6 into the active site of PI3K (PDB ID: 2RD0).
CONCLUSION
The research work embodied here is related to the in-silico prediction of the physicochemical and pharmacokinetic/ADME properties, drug-likeness, and molecular docking of some previously reported sarkomycin ester derivatives employing various online available servers. The predictive study unveiled that all the screened compounds 1-7 displayed excellent in-silico % absorption where the highest obtained value was 94.04%. The screened compounds have displayed high GI absorption and are predicted to be CNS active candidates as they can pass through the blood-brain barrier (BBB) except compound 1 which is CNS inactive. All the tested compounds are non-inhibitors of P-gp and Cytochrome P450 isomers except compound 7 which inhibits CYP1A2 and CYP2C19 and have low skin permeability. All the evaluated compounds were like a drug as they did not violate any of the considered rules except for compounds 2,3 and 4 which have one Muegge rule violation. They all have a bioavailability score of 0.55 and their drug-likeness and docking scores range from -1.13 to -0.30 and -5.1 to -6.2 respectively. The compound 6 has emerged to be the best in the in-silico predictions with a drug-likeness score of -0.30 without any rule violation and docking score of -6.2. In conclusion, these predictive evaluations give the guidance about the physico-chemical as well as pharmacokinetic/ADME traits along with enlightening the drug-likeness and binding/affinity pattern on the target protein of the tested compounds thus promoting the lead for the drug development program with additional efficacy.
REFERENCES


