International Journal of Pharmaceutical and Phytopharmacological Research
ISSN (Print): 2250-1029
ISSN (Online): 2249-6084

Tumor necrosis factor α may be a potent paracrine and endocrine mediator of inflammatory and immune functions. It is also known to manage the growth and differentiation of cells. The protein function of TNF-alpha is determined by its sequence and 3-D structure. The protein structures for seven different species were developed for "TNF alpha-induced protein”. in the absence of X-ray crystal structure and nuclear magnetic resonance (NMR) structure. In the present study, the 3-Dimensional molecular structures of seven “Tumor necrosis factor-alpha-induced proteins” from different species such as Homo sapiens (O95379), Xenopus tropicalis (Q5BKH4), Otolemur garnettii (B4UT01), Oryctolagus cuniculus (B7NZC7), Rattus norvegicus (Q6AYJ8), Mus musculus (Q9D8Y7), Callithrix jacchus (B0KWC3) were predicted by using Modeller10.1. The predicted model was then used to perform molecular docking simulations with ten different natural flavonoid derivatives to assess their ability. The docking results showed the lowest binding energies and good interaction for all seven modeled proteins.
INTRODUCTION
TNF alpha is derived from the tumor necrosis factor (TNF) superfamily that’s often realized as TNF; DIF; TNFA; TNFSF2; TNLG1F; cachectin. Initially, it was described in 1974 and cloned in 1984, recombinant TNF has been shown to induce the necrosis of transplanted methylcholanthrene-induced sarcomas in mice [1, 2]. TNF-α is a Protein Coding gene and a homotrimer with a subunit molecular mass of 17.3 kDa consisting of 157 amino acids [3] and in cell TNF is synthesized as a membrane-bound protein(26kDa/pro-TNF) which is released by TNFα converting enzyme (TACE, also known as ADAM-17) [4].
The encoding gene TNF-α is found in the region of the short arm of chromosome 6 between the HLA-B and HLA-DR genes in the major histocompatibility complex class III region [5]. This cytokine binds to TNFRSF1A/TNFR1 and TNFRSF1B/TNFBR [6]. It is mainly secreted by various types of cells including macrophages, monocytes, neutrophils, T cells, and NK cells [7-9].
TNF-α is a proinflammatory cytokine that leads to inflammation and immune responses. This multifunctional cytokine participates in numerous biological processes, pathological states [10] and plays an important role in the regulation of growth, differentiation, cell proliferation [11], inflammation, autoimmune diseases, insulin resistance progression, diabetes development [12], lipid metabolism and coagulation [13]. TNF-α is also involved in septic shock, tumorigenesis, hemorrhagic necrosis of tumors [14], viral replication, infections together with bacterial, fungal, viral, and parasitic infections. Besides, this cytokine incorporates a variety of diseases such as tuberculosis, cancer, asthma, psoriasis, malaria, etc.
The development of tumor necrosis factor-alpha (TNF alpha) inhibitors has been one among the foremost active areas of drug development for the treatment of inflammatory disease, rheumatoid arthritis, over the past decade. Pathways for therapeutic interventions and progressive steps are being established to improve the clinical effectiveness of anti-TNF-alpha strategies [15].
TNF-α plays a role in the regulation of several signaling pathways via two distinct receptors TNFR1 and TNFR2 [7, 16] which have been already mentioned above. The regulation of these receptors acts differently on various cell types both in normal and diseased tissue [17]. The predominant distinction among the two receptors in the death domain (DD) of TNFR-1 is absent in TNFR-2. For this reason, TNFR1 is an important member of the death receptor family, which can induce apoptotic cell death [18]. The expression of TNFR1 is on all cell types except erythrocyte, whereas the TNFR-2 is expressed mainly by immune cells [19]. Both apoptotic and pro-inflammatory are the signaling tend by TNFR-1, the suppression of these signaling is important for the autoimmune disease treatment. where TNFR-2 signaling is anti-inflammatory and cell proliferation and it promotes tissue repair and angiogenesis [20].
TNF is a pleiotropic protein [21] It is currently used in cancer treatment for soft tissue sarcoma (STS), irresectable tumors of various histological types, and melanoma in-transit metastases confined to the limb in the isolated limb perfusion (ILP) setting [22]. TNF- was found to be overexpressed in several neoplastic diseases, including prostate cancer, ovarian cancer, liver cancer, and breast cancer [23-26].
Approximately one million people worldwide are being treated or have been treated with TNF inhibitors available on the market, which include indications such as rheumatoid arthritis, psoriatic arthritis, psoriasis, and inflammatory bowel diseases, as well as a variety of potential clinical applications that are currently being evaluated at various stages [27-30].
TNF is produced by a variety of cell types, the most common of which being activated T cells and macrophages [31] TNF is generated as a transmembrane protein (tmTNF) that forms a homotrimer on the cell surface and can be cleaved into a soluble form (sTNF) by a metalloproteinase (TNF- convertase (TACE)) [5]. Both sTNF and tmTNF bind to TNF receptors 1 and 2, triggering a cascade of intracellular signaling events that result in the transcriptional activation of several pro-inflammatory genes [32].
In the present study, we constructed 3-dimensional protein structures for seven different “Tumor necrosis factor-alpha-induced protein 8” proteins with the use of Modeller, previously the selected proteins do not have any known crystal structure and also performed molecular docking studies with the use of Autodock. The homology model of these proteins was established using Modeller 10.1 and validated by using Procheck. Molecular docking studies were also performed on these modeled proteins with selected natural compounds by using Autodock 4.2.
MATERIALS AND METHODS
The amino acid sequences of all the seven “Tumor necrosis factor-alpha-induced protein 8” proteins from different species Homo sapiens (O95379), Xenopus tropicalis (Q5BKH4), Otolemur garnettii (B4UT01), Oryctolagus cuniculus (B7NZC7), Rattus norvegicus (Q6AYJ8), Mus musculus (Q9D8Y7), Callithrix jacchus (B0KWC3) were retrieved from UniprotKB (www.uniprot.org) [33] database. Initially, a sequence similarity search was performed by using the BLASTP tool [34] to all these protein sequences to find the related template proteins (crystal structures) by selecting the database as PDB [35]. Template structures were selected based on E-value, Identity, gaps, etc. ClustalX was used for the correction of alignment between query and template. Modeller10.1 was used to develop the models individually. Procheck was used to validate the modeled structures.
To find related protein templates to build models for these primary sequences, a sequence similarity search has been carried out separately by using the Protein BLAST tool against solved protein structures deposited in Protein Data Bank (PDB). ClustalX and ClustalW2 are used for the correction of alignment [36]. MODELLER 10.1 [37] was used to gain satisfactory models. MODELLER is an implementation of an automated approach to comparative modeling by satisfaction of spatial restraints which employs position-dependent gap penalties based on structural information of the template for generating alignments. After manually modifying the alignment input file in MODELLER 10.1 to match the template and query sequence, 20 models were generated. After the backbone generation, all the models were checked to select the protein with the least modeler objective function to validate. Backbone conformation was evaluated by the inspection of the psi/phi Ramachandran plot obtained from PROCHECK [38] analysis.
Docking studies
Molecular docking studies were performed to elucidate the binding mode of TNFAIP8 proteins with selected natural compounds. Autodock4.2 [39] was used for docking studies with modeled proteins. Autodock uses Lamarckian Genetic Algorithm. A total of ten natural compounds were selected for the docking study. All these compounds were downloaded (.sdf format) from the NCBI PubChem database. The compounds were loaded in Sybyl 6.7 [40] and were minimized by adding Gasteiger-Huckel charges and finally saved these molecules in. mol2 format. The seven modeled “Tumor necrosis factor-alpha-induced protein” proteins were docked separately. Initially, the protein was loaded and hydrogens were added to satisfy the valency before saving it in PDBQT format. The ligands were then loaded and conformations were set and it was saved in PDBQT format. X, Y, Z Coordinates were selected based on the amino acids present in the active site predicted in the Sybyl 6.7 biopolymer module. For all the dockings, a grid-point spacing of 0.375 Å was applied and a grid map with 60×60×60 points was used.
RESULTS AND DISCUSSION
Protein structure prediction: The current study reports that the Homology modeling studies of seven “Tumor necrosis factor-alpha-induced protein 8” (TNFAIP8) proteins in the absence of crystal structures. All the protein sequences were taken from the uniprotKB database. Later, a sequence similarity study was performed by using NCBI protein BLAST. The selected template protein structures have a high degree of homology with all the seven proteins, were used as a template, and had a good atomic resolution of its crystal structure. Corrections in the alignment were performed by using clustalX. After correction twenty models were generated and selected protein with the least modeler objective function for validation. Procheck was used to validate the modeled protein. All the models showed >90% of amino acids in the core region.
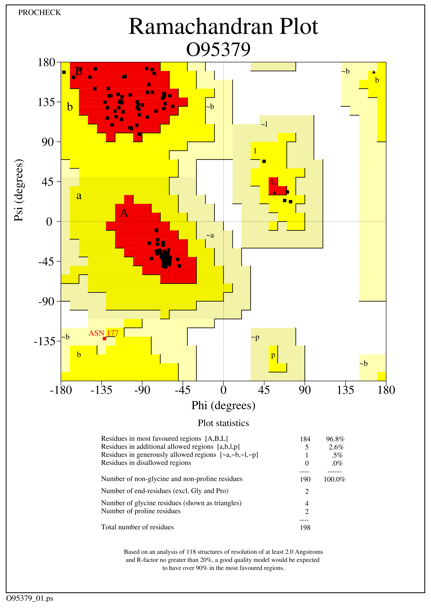
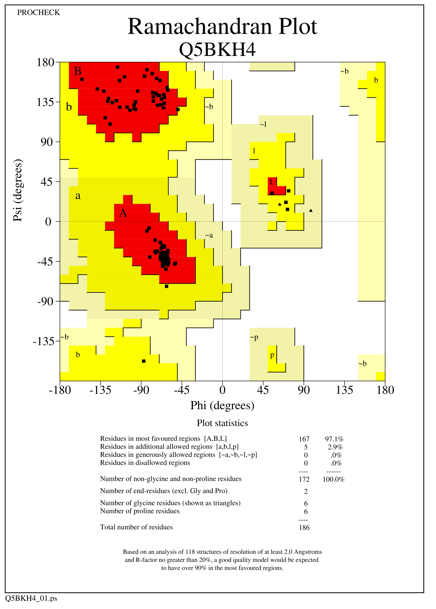
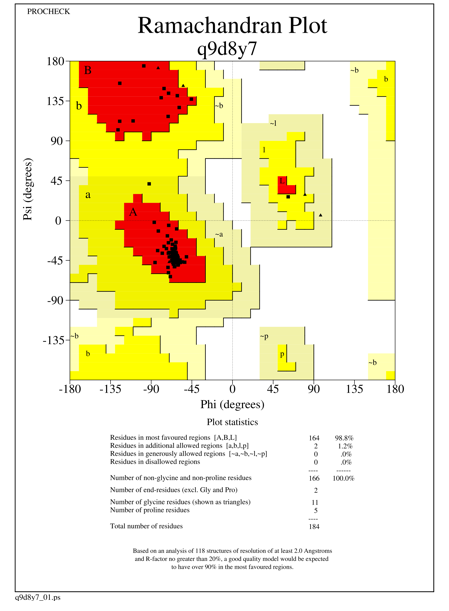
The generated models showed more than 96.0% of amino acid residues in the most favored region, 2-5% of amino acid residues in the additionally allowed region, with one amino acid residue present in generously allowed region for the species Homo sapiens, Rattus norvegicus and there are no amino acid residues present in the disallowed region. For Homo sapiens and Xenopus tropicalis we used 3F4M as a template, which constitutes 97.8% of the aa residues in the most favored region, 2.2% of the aa residues in additionally allowed region and there are no aa residues in generously and disallowed region. Template 4Q9V was used for the other five species which shows 93.8% (331 amino acid residues) of amino acids in the core region, 5.9% of amino acid residues (21 aa residues) in the additionally allowed region, one amino acid residue present in generously allowed region and there are no amino acid residues in the disallowed region. The statistics of all the seven models are given in Table 1 and Ramachandran plot statistics of O95379 (Homo sapiens) Q5BKH4 (Xenopus tropicalis) and Q9D8Y7 (Mus musculus) are shown in Figures 1-3.
Table 1. % of amino acid Residues falling in different regions of Ramachandran plot
|
S. No |
Name of the species |
Core region |
Allowed region |
Generously allowed region |
Disallowed region |
||||
|
No of residues |
% |
No of residues |
% |
No of residues |
% |
No of residues |
% |
||
|
1 |
Homo sapiens (O95379) |
184 |
96.8 |
5 |
2.6 |
1 |
0.5 |
0 |
0 |
|
2 |
Xenopus tropicalis (Q5BKH4) |
167 |
97.1 |
5 |
2.9 |
0 |
0 |
0 |
0 |
|
3 |
Otolemur garnettii (B4UT01) |
165 |
98.2 |
3 |
1.8 |
0 |
0 |
0 |
0 |
|
4 |
Oryctolagus cuniculus (B7NZC7) |
161 |
97.6 |
4 |
2.4 |
0 |
0 |
0 |
0 |
|
5 |
Rattus norvegicus (Q6AYJ8) |
166 |
98.2 |
2 |
1.2 |
1 |
0.6 |
0 |
0 |
|
6 |
Mus musculus (Q9D8Y7) |
164 |
98.8 |
2 |
1.2 |
0 |
0 |
0 |
0 |
|
7 |
Callithrix jacchus (B0KWC3) |
162 |
98.2 |
3 |
1.8 |
0 |
0 |
0 |
0 |
|
|
|
Figure 1. Ramachandran plot of the modeled Tumor necrosis factor-alpha-induced protein species Homo sapiens |
|
|
|
Figure 2. Ramachandran plot of the modeled Tumor necrosis factor-alpha-induced protein species Xenopus tropicalis |
|
|
|
Figure 3. Ramachandran plot of the modeled Tumor necrosis factor-alpha-induced protein species Mus musculus |
Molecular docking results
The most extensively used method for the calculation of protein-ligand interactions is Molecular docking. It is an efficient method to predict the potential ligand interactions. The present study uses secondary metabolites of native plants which have been identified as potent Tumor necrosis factor-alpha-induced protein inhibitors. The best binding conformation is assigned by the binding free energy assessment through AutoDock4.2 which uses a genetic algorithm. In total, ten natural compounds were docked against modeled Tumor necrosis factor-alpha-induced protein. After completion of the docking study complex files was generated for the lowest binding energy compounds and interactions were taken by using a DS visualizer.
However, the compound 4-Dehydrowithaferin A exhibited better interactions with five species of B4UT01 (Otolemur garnettii), B7NZC7 (Oryctolagus cuniculus), Q6AYJ8 (Rattus norvegicus), Q9D8Y7 (Mus musculus), B0KWC3 (Callithrix jacchus) modeled proteins, 3-Azeridinylwithaferin-A showed the lowest binding energy of -10.20 kcal/mol with interacting Arg125(2) and interactions with modeled O95379 (Homo sapiens) protein and Withanolide E exhibited lowest binding energy of -10.71 kcal/mol with interacting Phe111, Tyr105 for modeled Q5BKH4 (Xenopus tropicalis) protein. All the compounds have lower free energy values, indicating more thermodynamically favored interactions. Interactions and binding energies of all the natural compounds with seven modeled proteins are given in Table 2 and Figure 4 shows interactions of O95379 (Homo sapiens) Q5BKH4 (Xenopus tropicalis) and Q9D8Y7 (Mus musculus).
Table 2. Lowest Binding energy showing compounds with different species of Tumor necrosis factor-alpha-induced proteins
|
Uniprot protein id |
PubChem compound id with compound name |
Protein and ligand interaction |
Binding energy ΔG (Kcal/Mol) |
Dissociation Constant (kI) |
|
Q5BKH4 |
301751 Withanolide E |
Phe111, Tyr105 |
-10.71 |
14.1nM |
|
B4UT01 |
165541 4-Dehydrowithaferin A |
(Arg91)2 |
-7.44 |
3.35uM |
|
B7NZC7 |
165541 4-Dehydrowithaferin A |
Ala113 |
-8.90 |
301.46nM |
|
Q6AYJ8 |
165541 4-Dehydrowithaferin A |
Arg91 |
-6.50 |
17.17uM |
|
O95379 |
433929 3-Azeridinylwithaferin-A |
(Arg125)2 |
-10.20 |
33.4nM |
|
Q9D8Y7 |
165541 4-Dehydrowithaferin A |
Arg167, Thr110 |
-10.81 |
11.94nM |
|
B0KWC3 |
165541 4-Dehydrowithaferin A |
Gln55, Arg58 |
-7.49 |
3.25uM |
|
|
|
Figure 4. Molecular docking interactions of modeled proteins with natural compounds |
CONCLUSION
The crystal structures were built by performing homology modeling using Modeller10.1. The modeled protein was affirmed using PROCHECK. The generated models showed more than 96.0% of amino acid residues in the most favored region. The generated model was then docked with ten natural compounds from the plant Withania somnifera. The natural compounds were noted to show better binding energies. Withanolide E exhibited the lowest binding energy of -10.71 Kcal/mol with interacting Phe111 and Tyr105. The study proves that naturally existing compounds are more potent for the inhibition of Tumor necrosis factor-alpha-induced protein.
Acknowledgments: None
Conflict of interest: None
Financial support: None
Ethics statement: None