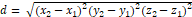International Journal of Pharmaceutical and Phytopharmacological Research
ISSN (Print): 2250-1029
ISSN (Online): 2249-6084

Structural computer-aided design of drugs is an effective modern way of creating targeted drugs. The essence of the method is to use intermolecular docking programs to select a ligand with a high affinity for the target protein. In the present study, we used the example of the search for ligands for the nonselective cationic channel TRPM8 to propose a two-step strategy based on deep neural networks and further verification by intermolecular docking. The strategy consists of using a neural network to screen out potential drug candidates and thereby reduce the list of candidate ligands for verification by intermolecular AutoDock program, which allows assessing the protein's affinity for the ligand by the minimum binding energy and identifying possible ligand conformations upon binding to certain centers of the protein, namely Y745 (Tyr 745 - critical center for TRPM8), R1008 (Phe 1008), and L1009 (Ala 1009). 8 from the ten potential ligands predicted by the neural network revealed minimum binding energy and a greater number of conformations in comparison to the classic TRPM8 ligand, menthol when verified by the AutoDock program. 2 ligands failed to dock, which may be due to insufficient allocated memory of the compute for successful docking or other technical problems. The proposed strategy is universal and will accelerate the search for ligands for various proteins.
INTRODUCTION
Structural computer-aided design of drugs is widely used now for creating targeted drugs [1]. The method consists of exploiting intermolecular docking programs to select a ligand with a high affinity for the target protein [2]. Using the example of the search for ligands for the nonselective calcium channel TRPM8 we propose a two-step strategy based on the initial search with a help of deep neural networks with further verification by molecular docking. TRPM8 is a member of transient receptor potential channels (TRP) important in sensory physiology. The results of a number of studies suggest that this receptor is associated with cellular response to low temperatures [3, 4]. A high level of TRPM8 expression in patients with asthma and COPD indicates its close relationship with the development of diseases of the respiratory system [5-7]. There are also certain indications of the possible role of the protein in the pathogenesis of prostate tumors [8, 9]. So, TRPM8 may be a potential target for drugs, created with a help of computer design [3, 10]. In this study, we tried to develop a new two-step strategy for the search for potential ligands for TRPM8. In the first step, we created a hybrid architecture neural network capable to accept receptor-ligand and classify them into interacting/non-interacting, thereby reducing the list of candidates for verification by a molecular docking program. After that, we tested the ability of the predicted ligands to form complexes with TRPM8 with a help of the AutoDock program.
MATERIALS AND METHODS
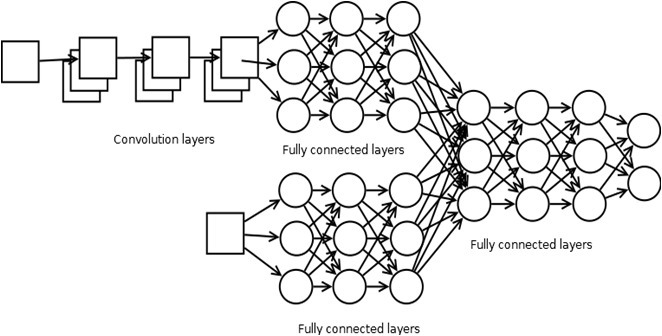
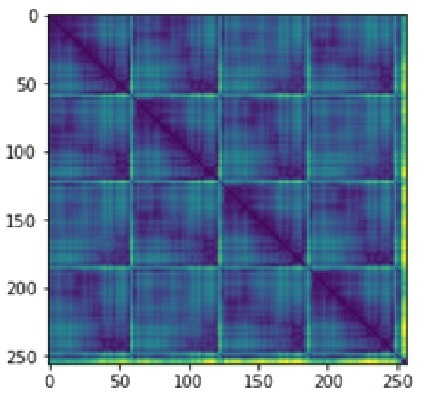
In a preselected two-step strategy, the first step is implementing a neural network to decrease the number of potential ligands for a target molecule. We developed a hybrid architecture for our neural network such that we could pass encoded receptor-ligand pairs simultaneously through layers (Figure 1). To represent every receptor structure interpolated distance matrix 256×256 was used, for every ligand smaller distance matrix 30×30 was used (Figure 2).
|
|
|
Figure 1. Hybrid neural network diagram |
|
|
|
Figure 2. Interpolated distance matrix for TRPM8 |
Protein tertiary structure can be represented via a unique 2D distance matrix reducing the computational complexity of further calculations. Distance between two atoms here is the distance between 2 points in the coordinate plane:
|
d= (x2-x1)2(y2-y1)2(z2-z1)2 |
(1) |
The whole dataset contains separated .pdb files with atom coordinates for each molecule. All coordinate files with annotations were downloaded from the BioLiP database [11, 12]. After that, according to the annotation file, receptor-ligand pairs were formed and represented as distance matrices and transformed into 2D tensors, since the deep learning framework PyTorch [13] used in this work requires tensors for computations. The neural network takes pair of tensors as input, receptor goes through following layers: Conv2d(1-32)->max_pool(10, 10)->Conv2d(32-64)->fc(4608-1024)->fc(1024-512)->fc(512-64), at the same time, tensor matrix goes through fc(900-512)->fc(512-128)->fc(128-64)->fc(64-64), at this step both tensors concatenates into a single one and pass through next layers: fc(128-64)->fc(64-64)->fc(64-2). Here, PyTorch notation is used, Conv2d means 2d convolution with the number of input-output features in parenthesis, fc means fully connected layers with the number of input-output neurons in parenthesis, max_pool means max pooling operation with kernel size in the parenthesis. Two output neurons represent the following "pair can interact" and "pair cannot interact". For each neuron, except the output two ReLU activation function was utilized, for output neurons SoftMax activation function was used. Mean square error was used as loss function and Adadelta with learning rate 0.5, rho 0.9, epsilon 10-6. Weights decay 0 was used as an optimization algorithm. Due to limitations in computational power, the network was trained for 500 epochs, on the training set with 884 pairs with batch size 128. After, we prepared the following dataset: TRPM8 and 98 ligands to check which of these can be classified via network as interacting pairs. Pairs that the network considered as interacting pairs were tested via AutoDock software [14].
Docking of TRPM8 with candidates for the role of ligands for this protein predicted by the neural network was performed using the MGLtools graphic molecular laboratory toolkit [15] and special software for molecular docking AutoDock. The TRPM8 3D structure in pdb file format (structure 6O6A) [16] was taken from the Research Collaboratory for Structural Bioinformatics (RCSB) resource [17]. The PubChem resource [18-20] was used to find the 2D structures of the ligands proposed by the neural network. Tyrosine 745 (Y745) hydroxy group was used as the binding point of the canonical TRPM8 agonist menthol [21, 22]. X:182.677; y:134.094; z:224.764 were chosen as the coordinates of the docking point. Docking was also carried out with Phe 1008 (R1008) and Alan 1009 (L1009) since these amino acid residues can also act as centers for the binding of some regulators [23, 24]. The coordinates were x:199.941; y:131.155; z:201.831 (for R1008) and x:201.618; y:134.349; z:199.033 (for L1009). According to the protocol, docking was carried out only with subunit B of TRPM8. Three other subunits - A, C, and D were removed. To increase the reactivity, the molecule was dehydrated and hydrogenated. Next, the studied ligand was added and meshes were applied to the desired area, namely, the areas where Y745, R1008, and L1009 were located. After overlaying the mesh, we indicated the selection of 5 positions and started docking. The result of docking was the obtained protein-ligand complex in the form of a dlg file with the number of stable conformations from the initial ones and the minimum binding energy. The obtained results were compared with the results of docking of the canonical TRPM8 agonist menthol.
RESULTS AND DISCUSSION
When using the neural network we created for classifying ligand/receptor pairs, the accuracy reached 70%. This is not a very high value, but it shows the potential of our method, and when using large computing power, the prediction accuracy can be increased. The description of 3D structures proposed in this work in a 2D form by the distances between each element seems to be a convenient and promising way for describing complex structures. The neural network suggested 10 ligands for TRPM8. Namely, gibberellin (A17), flavinadenine dinucleotide (FAD), dichlorophenylarsine (FDA), G4M, progesterone (57-83-0), aldosterone (52-39-1), goserelin (65807-02-5), xylometazoline (526-36-3), cortisol (53-06-5), and dexamethasone (III). Numerous molecules in the BioLiP base are presented in various forms and often only as a single domain. This seems to explain the fact that we were able to match only a small number of pairs. Below we give the names of the ligands without specifying a specific domain. The adequacy of the predictions of ligand for TRPM8 by the neural network was estimated with a help of molecular docking using the AutoDock software. As a result, eight out of ten stable complexes were identified at each center. The number of stable conformations and the established values of the minimum docking energy are shown in Tables 1-3.
Table 1. Minimum binding energies for the identified conformations of ligands when docked with the center Y745.
|
Ligands |
Minimum binding energy (kcal/mol) |
||||
|
Revealed conformations of the ligand |
|||||
|
1 |
2 |
3 |
4 |
5 |
|
|
A17 |
-4.4 |
-4.1 |
- |
- |
- |
|
FAD |
-4.7 |
-4.7 |
-4.7 |
-4.7 |
-1.9 |
|
G4M |
-9.5 |
-8.4 |
-8.4 |
-5.4 |
-4.8 |
|
57-83-0 |
-5.5 |
- |
- |
- |
- |
|
52-39-1 |
-8.7 |
-6.9 |
- |
- |
- |
|
526-36-3 |
-5 |
- |
- |
- |
- |
|
53-06-5 |
-8 |
- |
- |
- |
- |
|
III |
-5.7 |
-5.6 |
- |
- |
- |
Table 2. Minimum binding energies for the identified conformations of ligands when docked with the center R1008.
|
Ligands |
Minimum binding energy (kcal/mol) |
||||
|
Revealed conformations of the ligand |
|||||
|
1 |
2 |
3 |
4 |
5 |
|
|
A17 |
-5.8 |
-5.6 |
-5.6 |
- |
- |
|
FAD |
-11.9 |
-10 |
-9 |
-8.3 |
-5 |
|
G4M |
-10 |
-9.6 |
-7.6 |
-6.5 |
-3.4 |
|
57-83-0 |
-6.3 |
- |
- |
- |
- |
|
52-39-1 |
-8.8 |
-8.6 |
- |
- |
- |
|
526-36-3 |
1.-5.18 |
-4.9 |
- |
- |
- |
|
53-06-5 |
1.-8.40 |
-8.2 |
- |
- |
- |
|
III |
1.-9.62 |
-9.3 |
- |
- |
- |
Table 3. Minimum binding energies for the identified conformations of ligands when docked with the center L1009.
|
Ligands |
Minimum binding energy (kcal/mol) |
||||
|
Revealed conformations of the ligand |
|||||
|
1 |
2 |
3 |
4 |
5 |
|
|
TablesA17 |
-5.5 |
-5.4 |
-5.0 |
4.- |
5.- |
|
FAD |
-12 |
-10.4 |
-6.7 |
-3.4 |
- |
|
G4M |
-10.3 |
-9.3 |
-9.1 |
-9.1 |
-7.1 |
|
57-83-0 |
-5.6 |
- |
3.- |
4.- |
5.- |
|
52-39-1 |
-9.2 |
-8 |
3.- |
4.- |
5.- |
|
526-36-3 |
-5.6 |
-5.4 |
-5.4 |
- |
- |
|
53-06-5 |
-10.1 |
-8.7 |
-8.2 |
- |
- |
|
III |
-11.3 |
-10.1 |
- |
- |
- |
The minimum binding energy reflects the affinity of the receptor for the ligand. The lower it is, the more durable the protein-ligand complex. Most neural network-predicted ligands have lower binding energy (Tables 1-3) than the canonic agonist of TRPM8 – menthol, docked in the same coordinate as the predicted ligands (Table 4).
Table 4. The minimum binding energy of the identified conformations of menthol (ligand 1490-04-6) with centers Y745, R1008, L1009.
|
Binding centers |
Minimum binding energy (kcal/mol) |
|
|
Conformations |
||
|
1 |
2 |
|
|
Y745 |
-4.69 |
-4.45 |
|
R1008 |
-4.94 |
- |
|
L1009 |
-5.41 |
- |
Of the candidates suggested by the neural network, FAD and G4M revealed the largest number of conformations (5 out of 5) with centers R1008 and Y745. For the L1009 center, FAD revealed successful docking for 4 of 5 specified conformations. It should be concluded that it is these ligands that have the highest affinity for TRPM8. Two of the predicted ligands, namely FDA and goserelin, failed to dock. AutoDock software is unable to recognize the arsenic atom in the FDA, and because of this FDA failed to dock with the centers Y745, R1008, and L1009.
he reason for the error for docking may be the damaged source files of the ligand. pdbqt, or the insufficient allocated memory of the computer. The unsuccessful docking of goserelin with the centers may be explained by the large size of the ligand. It may be the reason for the error at each docking attempt. The explanation of the program for the unsuccessful docking consist of numerous mobile bonds and C atoms. The proposed two-step strategy for the search for ligands for TRPM8 is universal. It allows quickly test the affinity of a large number of possible ligands for a particular protein molecule. After improvement, our approach may significantly facilitate the search for ligands for any protein. It should be noted that the established ligand candidates are capable of forming a stable complex with the receptor. However, whether they are agonists or antagonists for TRPM8 is unknown. To answer this question, experiments on cell cultures or on model animals are required. Four from the ligands predicted by the neural network that form complexes with TRPM8 are steroid hormones. Drugs based on corticosteroids have long been used as anti-inflammatory and immunosuppressive drugs. As mentioned above TRPM8 is involved in the formation of bronchial hypersensitivity to cold in patients with bronchial asthma [4, 25-27]. The high affinity of corticosteroids to TRPM8 established in this work can explain their pharmacological effects.
The proposed strategy based on a combination of the use of neural networks and molecular docking needs more computing power than was available to us. Therefore, the dataset we used for our project was insufficient. The method will be refined in the future and will allow a more accurate search for candidates for the role of a drug with a high affinity for receptor molecules.
CONCLUSION
In the present study, we proposed a strategy for predicting potential ligands for TRPM8 in silico, based on the use of machine learning tools based on deep neural networks and further verification by intermolecular docking. The use of a neural network makes it possible to prescreen potential drug candidates and thereby reduce the list of candidate ligands for verification using intermolecular docking (AutoDock), which evaluates the affinity of the protein for the ligand by the minimum binding energy and reveals possible ligand conformations upon binding to certain amino acid residues of the protein. The latter were used: Tyr 745, Phe 1008, and Ala 1009. Of the ten ligands, which were suggested by the neural network, eight revealed high minimum binding energy and more conformations in relation to the classic TRPM8 ligand menthol, when verified by the AutoDock program. The two suggested ligands did not show the interaction with TRPM8, which may be due to some technical problems. The proposed strategy is universal and will speed up the search for ligands for various proteins.
Acknowledgments: None
Conflict of interest: None
Financial support: None
Ethics statement: None